選對路徑是成功關鍵:三大途徑適用情境一次看懂
FDA 醫療器械上市路徑選擇取決於「產品風險分類」與「是否有同類產品(Predicate Device)」。根據 21 CFR Part 807 與 21 CFR Part 814 規定,主要有三大審查途徑:
- 510(k) Premarket Notification:適用於 Class II 或部分 Class I 器械,需證明與已核准產品「實質等效」(Substantially Equivalent)
- De Novo Classification:適用於低至中風險但無 Predicate Device 的創新器械,可建立新產品分類
- PMA (Premarket Approval):適用於 Class III 高風險器械,需提供完整臨床數據證明安全性與有效性
選錯路徑的代價: 若選擇 510(k) 但 FDA 判定需 PMA,需重新申請並補充臨床數據,可能延誤 1-3 年上市時程。反之,若可走 510(k) 卻選擇 PMA,將大幅增加成本(PMA 費用約 510(k) 的 10-20 倍)與審查時間。
若選擇 510(k) 但 FDA 判定需 PMA,需重新申請並補充臨床數據,可能延誤 1-3 年上市時程。反之,若可走 510(k) 卻選擇 PMA,將大幅增加成本(PMA 費用約 510(k) 的 10-20 倍)與審查時間。
FDA 三大審查途徑完整比較
核心差異總覽表
| 比較項目 | 510(k) | De Novo | PMA |
|---|---|---|---|
| 適用分類 | Class II(部分 Class I) | Class I/II(無 Predicate) | Class III |
| 核心要求 | 證明 Substantial Equivalence | 證明低至中風險 + 無 Predicate | 完整臨床數據 + 安全有效性 |
| 審查時間 | 90 天(FDA 目標,實際 3-6 個月) | 150 天(FDA 目標,實際 6-12 個月) | 180 天(FDA 目標,實際 1-3 年) |
| 申請費用 | $13,000-$20,000(2026 MDUFA) | $122,000-$150,000 | $400,000-$500,000+ |
| 臨床數據 | 通常不需要(除非 FDA 要求) | 可能需要(視風險而定) | 必須提供 |
| 核准後監管 | Post-Market Surveillance | Post-Market Surveillance | 嚴格 Post-Approval Study |
| 成功率 | 約 85-90% | 約 75-80% | 約 50-60% |
⚠️ 重要提醒:費用為 2026 年 MDUFA V 標準,實際費用視公司規模(Small Business 可減免)與產品複雜度而異。
圖 2: FDA 醫療器械上市路徑選擇決策樹
圖片用途說明:
此決策樹將「510(k) vs De Novo vs PMA」的選擇邏輯視覺化,幫助台灣醫材廠快速判定適合的審查途徑。
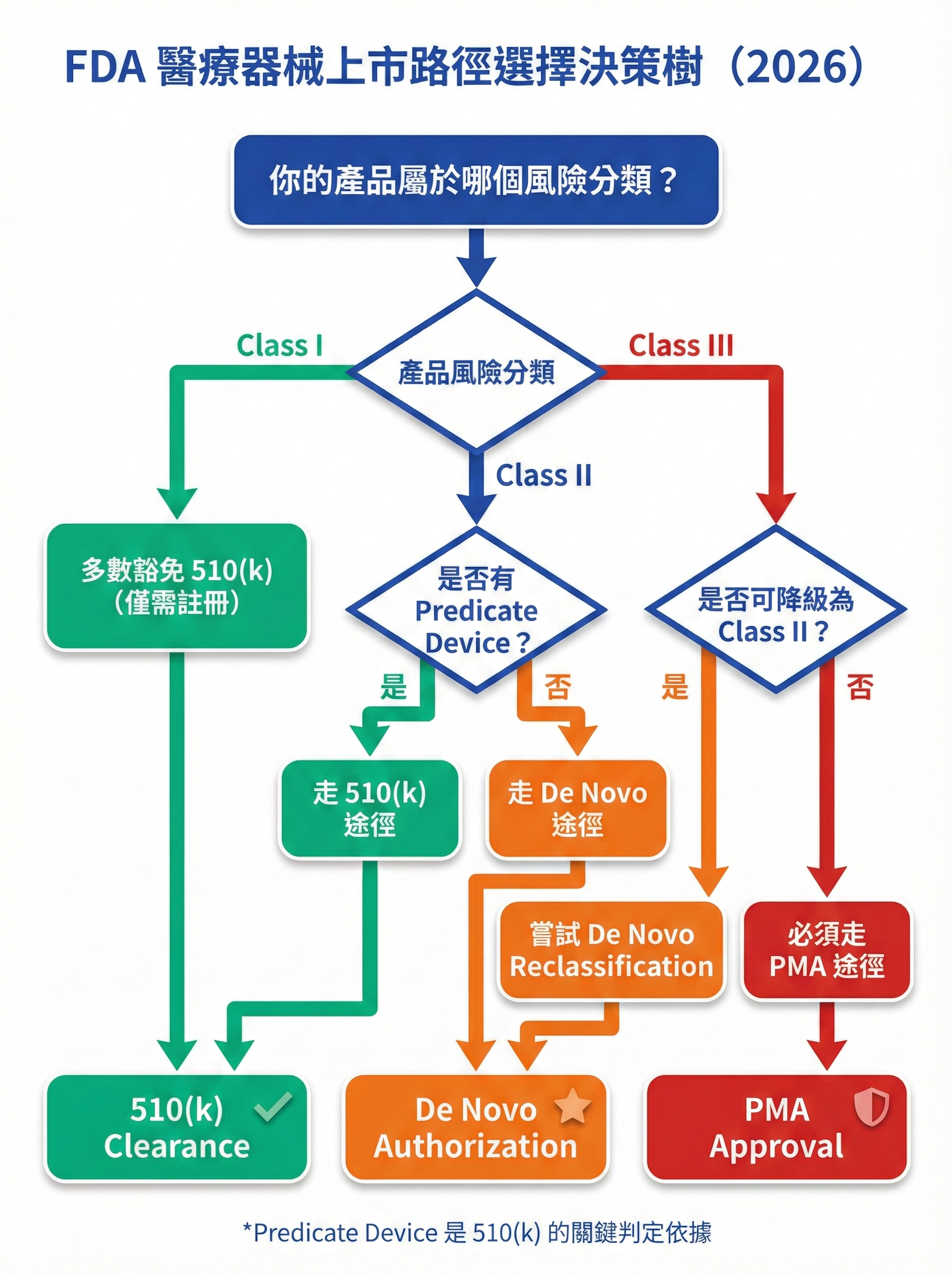
510(k) 途徑:最常見但需找到 Predicate Device
適用條件與核心要求
適用產品:
- Class II 醫療器械(約占所有器械的 43%)
- 部分 Class I 器械(若未豁免 510(k))
- 需與已核准的 Predicate Device 具「實質等效性」(Substantial Equivalence)
Substantial Equivalence 判定標準:
- Intended Use 相同或相似:預期用途必須一致
- 技術特性相同:材料、設計、能源來源、操作原理等
- 性能數據相當:若技術特性不同,需證明性能不劣於 Predicate Device
申請流程:
- 確認產品分類(使用 FDA Product Classification Database)
- 尋找 Predicate Device(使用 FDA 510(k) Database)
- 準備 510(k) 申請文件(包含 Substantial Equivalence 比較、性能測試數據)
- 提交至 FDA(電子提交系統 eSTAR)
- FDA 審查(90 天目標,可能要求補件)
- 獲得 510(k) Clearance
💡 實務技巧:選擇 Predicate Device 時,優先選擇「近期核准」(5 年內)且「技術特性高度相似」的產品,可降低 FDA 要求額外數據的風險。
常見退件原因與避坑指南
退件原因 Top 3:
- Predicate Device 選擇不當:Intended Use 不一致或技術差異過大
- 性能測試數據不足:未提供足夠的 Bench Testing 或 Biocompatibility 數據
- 標籤資訊不完整:未符合 21 CFR Part 801 要求
避坑策略:
- 提交前進行 Pre-Submission Meeting(與 FDA 討論 Predicate Device 選擇)
- 委託第三方實驗室進行性能測試(確保數據可信度)
- 參考 FDA Guidance Documents(如 “The 510(k) Program: Evaluating Substantial Equivalence”)
De Novo 途徑:創新器械的新選擇
適用條件與核心優勢
適用產品:
- 低至中風險器械(Class I 或 Class II)
- 無 Predicate Device 可供比較(全新類型產品)
- 可證明產品風險可透過 General Controls 或 Special Controls 管理
核心優勢:
- 建立新產品分類:成功後可成為其他廠商的 Predicate Device
- 避免 PMA 高成本:相較 PMA 節省 60-80% 費用與時間
- 市場先機:成為該類產品的「First in Class」
申請流程:
- 確認產品無 Predicate Device(查詢 FDA 510(k) Database)
- 評估產品風險等級(是否可透過 General/Special Controls 管理)
- 準備 De Novo 申請文件(包含風險分析、性能數據、建議 Controls)
- 提交至 FDA(電子提交系統)
- FDA 審查(150 天目標,可能要求補件或臨床數據)
- 獲得 De Novo Authorization 與新產品分類
⚠️ 重要提醒:De Novo 成功率約 75-80%,若被拒絕,可能需改走 PMA 途徑或重新設計產品降低風險。
典型成功案例
案例一:智慧手環心率監測功能
- 產品:具心率監測功能的智慧手環
- 挑戰:無 Predicate Device(當時智慧手環尚未被分類為醫療器械)
- 解決方案:透過 De Novo 建立新分類(Class II, Special Controls)
- 結果:成為後續同類產品的 Predicate Device
案例二:AI 輔助診斷軟體
- 產品:AI 影像辨識輔助診斷軟體
- 挑戰:AI 演算法無傳統 Predicate Device
- 解決方案:De Novo 申請,提供演算法驗證數據與臨床性能評估
- 結果:建立 AI 醫療軟體新分類
PMA 途徑:高風險器械的嚴格審查
適用條件與挑戰
適用產品:
- Class III 高風險器械(如心臟支架、人工心臟瓣膜、植入式除顫器)
- 直接維持生命或預防嚴重健康損害的器械
- 需提供完整臨床數據證明「合理保證安全性與有效性」(Reasonable Assurance of Safety and Effectiveness)
核心挑戰:
- 臨床試驗成本高:需進行 IDE(Investigational Device Exemption)臨床試驗,成本可達數百萬美元
- 審查時間長:平均 1-3 年(含補件與臨床數據審查)
- 成功率較低:約 50-60%(嚴格的安全性與有效性標準)
申請流程:
- 進行臨床前測試(Bench Testing, Animal Studies)
- 申請 IDE(若需進行臨床試驗)
- 完成臨床試驗(需符合 GCP 與 21 CFR Part 812)
- 準備 PMA 申請文件(包含完整臨床數據、製造資訊、標籤)
- 提交至 FDA(電子提交系統)
- FDA 審查(可能召開 Advisory Committee Meeting)
- 獲得 PMA Approval
💡 實務技巧:PMA 申請前建議進行多次 Pre-Submission Meeting,與 FDA 確認臨床試驗設計、主要終點(Primary Endpoint)與統計分析計畫,可降低後續補件風險。
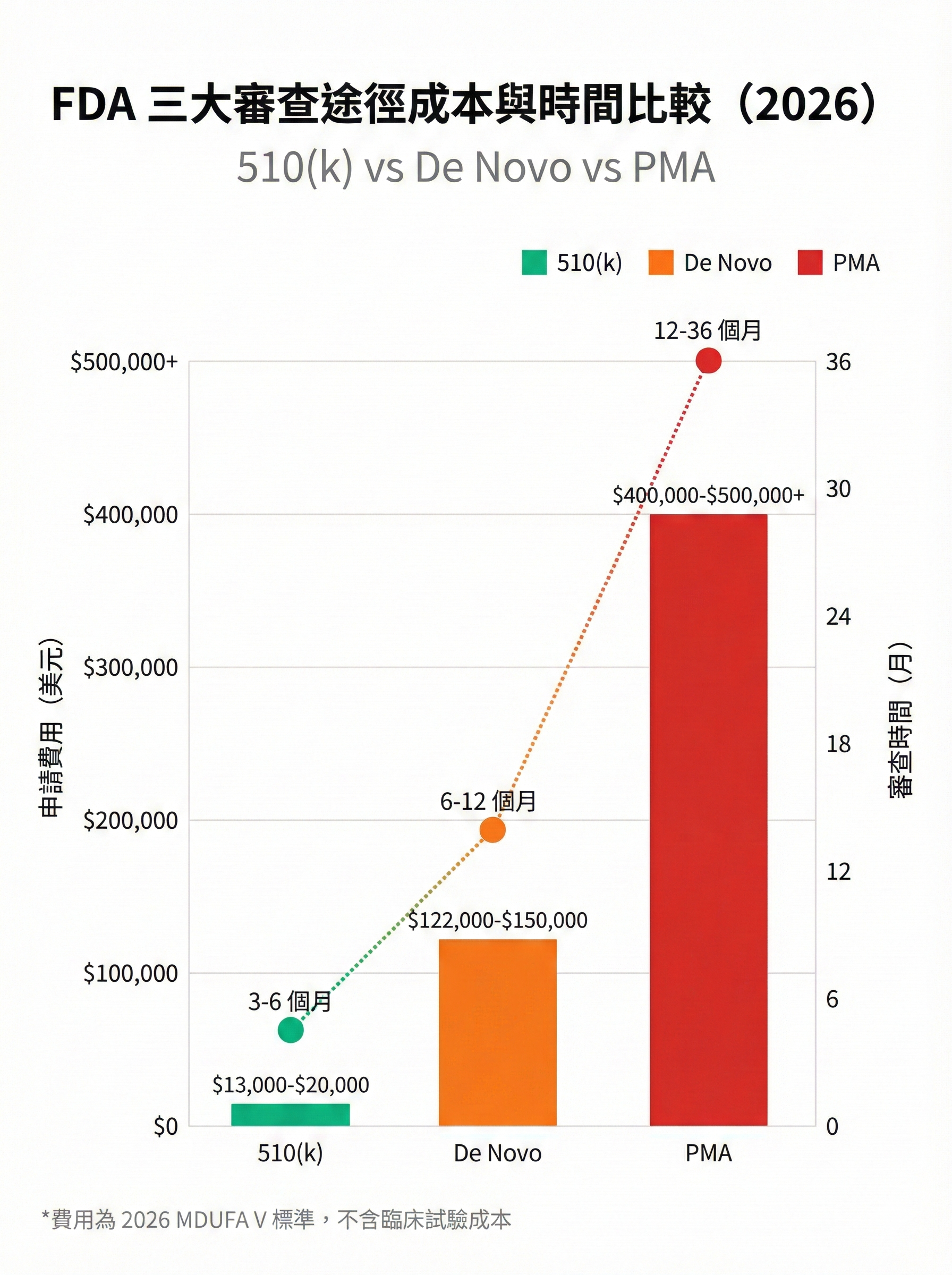
圖 3: FDA 三大審查途徑成本與時間比較
圖片用途說明:
此比較圖整理 510(k)、De Novo、PMA 三大途徑的成本與審查時間差異,幫助業者評估資源投入與上市時程規劃。
如何選擇最適合的審查途徑?
選擇策略決策框架
步驟一:確認產品風險分類
- 使用 FDA Product Classification Database 查詢
- 參考 21 CFR Part 862-892 產品分類規定
- 若無明確分類,可提交 513(g) Request(分類判定請求)
步驟二:評估 Predicate Device 可得性
- 搜尋 FDA 510(k) Database(至少找 2-3 個候選)
- 比較 Intended Use、技術特性、性能參數
- 若無合適 Predicate,考慮 De Novo 或 PMA
步驟三:評估資源與時程
- 510(k):適合資源有限、需快速上市的 Class II 產品
- De Novo:適合創新產品、願意投入中等資源建立新分類
- PMA:適合 Class III 產品、有充足資源進行臨床試驗
步驟四:考量長期策略
- 510(k):上市快但無專屬性(競爭者可用你的產品作 Predicate)
- De Novo:可建立市場先機,成為 Predicate Device
- PMA:核准後具高進入門檻,競爭者難以複製
需要特別注意的 3 個選擇陷阱
陷阱一:誤判產品風險分類
風險:自行判定為 Class II 並準備 510(k),但 FDA 審查後要求改走 PMA,導致重新申請與補充臨床數據。
避險策略:
- 提交前進行 513(g) Request(分類判定請求)
- 參加 Pre-Submission Meeting 與 FDA 確認分類
- 參考同類產品的核准歷史
陷阱二:選擇不當的 Predicate Device
風險:選擇的 Predicate Device 與新產品 Intended Use 或技術特性差異過大,導致 510(k) 被拒絕或要求補充大量數據。
避險策略:
- 選擇「近期核准」(5 年內)的 Predicate
- 優先選擇「技術特性高度相似」的產品
- 準備多個備選 Predicate(至少 2-3 個)
陷阱三:低估 De Novo 或 PMA 的時間與成本
風險:未充分評估 De Novo 或 PMA 的資源需求,導致申請過程中資金或時間不足,被迫中止或延遲。
避險策略:
- 進行詳細的資源規劃(費用、人力、時程)
- 預留 30-50% 緩衝預算(應對補件或額外測試)
- 考慮分階段融資或尋找策略合作夥伴
總結
關鍵重點:
- FDA 醫療器械上市路徑選擇取決於產品風險分類與 Predicate Device 可得性
- 510(k):最常見途徑,適合 Class II 產品且有合適 Predicate(成本低、時間短)
- De Novo:適合創新產品無 Predicate,可建立新分類與市場先機(中等成本與時間)
- PMA:適用 Class III 高風險產品,需完整臨床數據(成本高、時間長、門檻高)
- 選擇前務必確認產品分類、評估 Predicate 可得性、規劃充足資源
- 透過 Pre-Submission Meeting 與 FDA 確認策略,可大幅降低退件風險
若對 FDA 醫療器械上市路徑選擇仍有疑問,歡迎參考 綠圈圈官網 的專業顧問服務。