
品牌與工廠誰負責?FDA 明確規定責任主體
在 FDA 醫療器械法規下,責任歸屬取決於「誰向美國市場引入產品」(Who introduces the device into U.S. commerce)。根據 21 CFR Part 807 與 21 CFR Part 820,責任主體包含三大角色:
- 製造商(Manufacturer):實際生產器械的工廠,需符合 QSR(21 CFR Part 820)品質系統要求
- 規格開發者(Specification Developer):制定產品設計規格、Intended Use 的品牌商或委託方
- 美國進口商/經銷商(Initial Distributor/Importer):首次將產品引入美國市場的實體
關鍵判定原則:若品牌商僅委託代工但掌握產品設計、標籤與 Intended Use,仍可能被視為 Manufacturer 或 Specification Developer,需承擔主要合規責任。若代工廠自行設計產品並貼牌銷售,則代工廠為主要責任方。實務上,FDA 可能同時追究多方責任,形成「連帶責任」(Joint Liability)風險。
FDA 醫療器械法規下的三大核心角色
角色一:製造商(Manufacturer)的責任範圍
根據 21 CFR 820.3(o) 定義,製造商包含:
- 實際生產器械的工廠(含 ODM/OEM 代工廠)
- 重新包裝或重新標籤的經銷商
- 規格開發者(Specification Developer,即制定設計規格的品牌商)
主要責任:
- 完成 Establishment Registration(設施註冊)
- 完成 Device Listing(器械列表申報)
- 符合 QSR(21 CFR Part 820)品質系統要求
- 提交 510(k)、PMA 或 De Novo 申請(視產品分類)
- 確保產品標籤符合 21 CFR Part 801 要求
- 建立 UDI(Unique Device Identification)系統
⚠️ 重要提醒:即使是代工廠,只要實際生產器械,就必須完成 FDA 註冊與 Device Listing,並符合 QSR 要求。
角色二:規格開發者(Specification Developer)的責任陷阱
這是台灣品牌商最容易忽略的角色!
根據 21 CFR 820.3(o),規格開發者指「制定或建立器械規格的人或實體」,包含:
- 制定產品設計規格、功能要求
- 決定產品的 Intended Use(預期用途)
- 控制產品標籤與宣稱內容
責任陷阱:
- 即使未實際生產,品牌商若掌握設計規格與 Intended Use,仍可能被視為 Manufacturer
- 需與代工廠共同承擔 QSR 合規責任
- 若產品出現品質問題或違規,FDA 可同時追究品牌商與代工廠
💡 實務技巧:品牌商應在合約中明確約定「誰是 Manufacturer of Record」(註冊製造商),並確保該方完成所有 FDA 合規義務。
角色三:美國代理人(US Agent)與進口商責任
美國代理人(US Agent):
- 海外製造商必須指定的美國境內聯絡人
- 負責接收 FDA 通訊與檢查通知
- 不承擔產品合規責任,僅為聯絡窗口
初始經銷商/進口商(Initial Distributor):
- 首次將產品引入美國市場的實體
- 需完成 Establishment Registration(若從事重新包裝或重新標籤)
- 可能因「引入不合規產品」被 FDA 追究責任
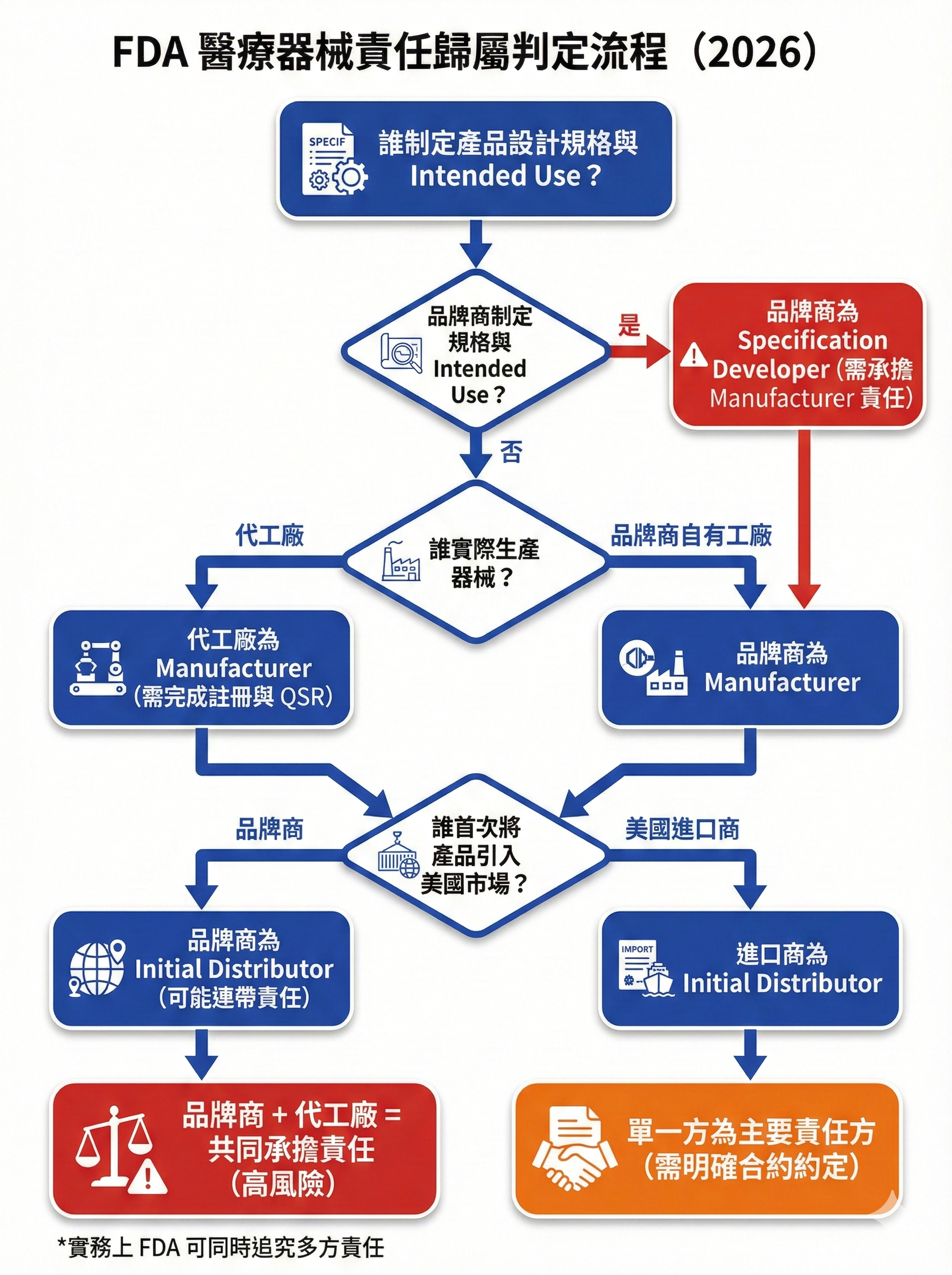
圖 2: FDA 醫療器械責任歸屬判定流程
圖片用途說明:
此決策樹將「品牌商 vs 製造商」責任歸屬的判定邏輯視覺化,幫助台灣業者快速判斷自身角色與責任。
5 大法律風險情境與避險策略
情境一:品牌商被視為 Specification Developer
風險:品牌商雖未實際生產,但因制定產品規格與 Intended Use,被 FDA 視為 Manufacturer,需承擔 QSR 合規責任。
避險策略:
- 在委託合約中明確約定「代工廠為 Manufacturer of Record」
- 確保代工廠完成 FDA 註冊、Device Listing 與 QSR 認證
- 品牌商仍需監督代工廠合規狀況(Design Control 責任)
情境二:代工廠未完成 FDA 註冊
風險:代工廠未完成 Establishment Registration 或 Device Listing,導致產品被視為「未註冊器械」,品牌商與代工廠可能同時被 FDA 警告。
避險策略:
- 出貨前確認代工廠已完成 FDA 註冊(可於 FDA 官網查詢)
- 要求代工廠提供 Registration Number 與 Device Listing 證明
- 合約中約定「未完成註冊不得出貨」條款
情境三:產品標籤不符合 FDA 要求
風險:產品標籤缺少 UDI、製造商資訊或使用說明,導致被 FDA 扣留或警告,品牌商與代工廠可能連帶受罰。
避險策略:
- 明確約定「誰負責標籤設計與合規審查」
- 確保標籤包含 21 CFR Part 801 要求的所有資訊
- 使用 FDA 認可的 UDI Issuing Agency(如 GS1、HIBCC)
情境四:產品出現品質問題或不良事件
風險:產品發生不良事件(Adverse Event),FDA 要求召回(Recall)或發布警告信(Warning Letter),品牌商與代工廠可能共同承擔責任。
避險策略:
- 建立 MDR(Medical Device Reporting)通報機制
- 合約中約定「品質問題責任分擔比例」
- 購買產品責任保險(Product Liability Insurance)
情境五:FDA 檢查發現 QSR 缺失
風險:FDA 對代工廠進行檢查,發現 QSR(21 CFR Part 820)缺失,品牌商若為 Specification Developer,可能被要求改善或停止銷售。
避險策略:
- 定期稽核代工廠 QSR 符合性(至少每年一次)
- 要求代工廠提供 ISO 13485 認證
- 建立 Supplier Quality Agreement(供應商品質協議)
圖 3: FDA 醫療器械法律風險等級矩陣
圖片用途說明:
此風險矩陣整理品牌商與代工廠在不同情境下的法律風險等級,幫助業者快速評估自身風險。

需要特別注意的 3 個合規紅線
紅線一:未指定 Manufacturer of Record
風險:品牌商與代工廠皆未於 FDA 註冊為 Manufacturer,導致產品被視為「無註冊製造商」,可能被扣留或禁止進口。
解決方案:
- 在委託合約中明確約定「誰為 Manufacturer of Record」
- 確保該方完成 Establishment Registration 與 Device Listing
- 若品牌商為 Specification Developer,需與代工廠共同承擔責任
紅線二:Intended Use 與實際宣稱不一致
風險:產品標籤或行銷宣稱超出原申報的 Intended Use,可能被 FDA 視為「未核准新用途」(Off-Label Use),導致警告信或召回。
解決方案:
- 所有宣稱必須與 510(k) 或 PMA 申報的 Intended Use 一致
- 避免在標籤、網站或社群媒體上宣稱未核准的功能
- 若需擴大用途,需提交新的 510(k) 或 PMA Supplement
紅線三:忽略 Post-Market 監督責任
風險:產品上市後,品牌商與代工廠未建立 MDR(Medical Device Reporting)機制,導致不良事件未通報,可能被 FDA 重罰。
解決方案:
- 建立 MDR 通報流程(30 天內通報嚴重不良事件)
- 定期進行 Post-Market Surveillance(上市後監督)
- 合約中約定「誰負責 MDR 通報」(通常為品牌商)
💡 實務技巧:建議品牌商與代工廠簽署「Quality Agreement」(品質協議),明確約定各方責任、稽核頻率與不良事件處理流程。
總結
關鍵重點:
- FDA 醫療器械責任歸屬取決於「誰制定規格」「誰實際生產」「誰引入美國市場」
- 品牌商若為 Specification Developer,即使未實際生產,仍可能被視為 Manufacturer
- 代工廠必須完成 FDA 註冊、Device Listing 與 QSR 合規
- 實務上 FDA 可同時追究品牌商與代工廠的連帶責任
- 透過合約明確約定責任分擔、建立稽核機制與購買保險,可降低法律風險
若對 FDA 醫療器械責任歸屬與法律風險仍有疑問,歡迎參考 綠圈圈官網 的專業顧問服務。