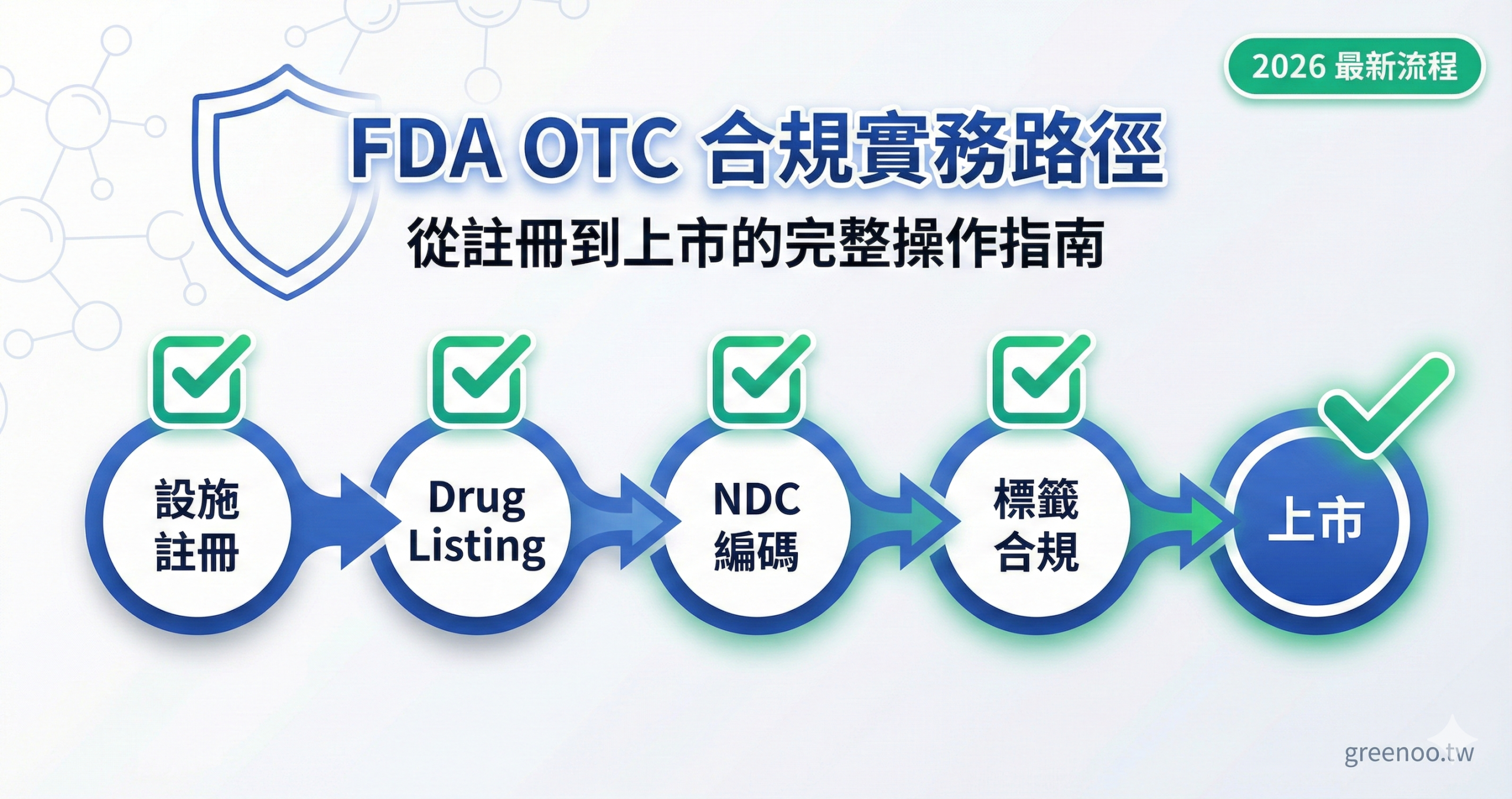
FDA OTC 合規到底要做哪些事?完整流程一次掌握
要將產品以 OTC 藥品身份在美國上市,必須完成四大核心步驟:設施註冊(Establishment Registration)、藥品列表申報(Drug Listing)、NDC 編碼取得,以及標籤合規審查。 這些步驟環環相扣,任一環節缺失或錯誤,都可能導致產品無法合法銷售,甚至面臨 FDA 執法行動。對台灣賣家與外貿工廠而言,最常見的問題是「不知道從哪裡開始」或「誤以為完成註冊就能上市」,實際上每個步驟都有明確的時程要求與文件規範。
本文將完整拆解 FDA OTC 合規的實務操作路徑,從第一步設施註冊到最後的標籤審查,提供具體的操作指引、常見錯誤與時程規劃,讓你一次看懂整個合規流程。
為什麼需要這份完整路徑指南?
許多台灣業者在接觸 FDA OTC 法規時,常因資訊分散而無法建立完整的合規藍圖。有些人誤以為「只要產品符合 Monograph 就能賣」,卻忽略了設施註冊與 Drug Listing 的強制要求;有些人則在申報 Drug Listing 時才發現 NDC 編碼規則複雜,導致申報失敗需重新來過。
根據 FDA 規定(21 CFR Part 207),所有在美國製造或進口的 OTC 藥品,其製造設施與產品資訊都必須向 FDA 申報。這不僅是法律要求,更是產品能否進入美國市場的先決條件。若未完成這些步驟就銷售產品,將被視為「未經核准藥品」(Unapproved Drug),面臨產品扣押、罰款,甚至刑事責任。
對於想深入了解 OTC 產品如何被判定為藥品的業者,建議先閱讀《台灣賣家與外貿工廠出口美國 OTC 合規全指南:你的產品是否已被視為藥品?》,釐清產品定位後再進入實務操作。
步驟一:設施註冊(Establishment Registration)- 取得入場資格
什麼是設施註冊?
設施註冊是指製造、包裝、貼標或持有 OTC 藥品的設施,必須在 FDA 的藥品設施註冊系統(DRLM – Drug Registration and Listing Module)中登記。這是 FDA 識別與追蹤藥品來源的基礎機制,也是後續 Drug Listing 的前提條件。
誰需要註冊?
- 製造商:實際生產 OTC 產品的工廠(包含台灣代工廠)
- 包裝商:進行二次包裝或分裝的設施
- 貼標商:負責產品標籤製作與黏貼的設施
- 美國進口商:若產品從海外進口,進口商也需註冊
⚠️ 常見誤區:許多台灣工廠誤以為「只有美國境內的設施需要註冊」,實際上海外製造設施同樣必須註冊,且需指定美國代理人(US Agent)。
註冊操作步驟
- 準備設施資訊:公司名稱、地址、聯絡人、營業執照等
- 指定美國代理人:必須是位於美國境內的個人或公司,作為 FDA 與設施的聯絡窗口
- 登入 FDA DRLM 系統:使用 FDA Industry Systems 帳號登入
- 填寫設施資訊:包含設施類型(製造/包裝/貼標)、產品類別、cGMP 狀態等
- 提交註冊:系統會立即生成註冊編號(Establishment Identifier)
時程與更新
- 初次註冊:可隨時進行,建議在產品上市前 3-6 個月完成
- 年度更新:每年 10 月 1 日至 12 月 31 日必須更新註冊資訊
- 變更通知:設施地址、所有權、美國代理人等重大變更需在 30 天內更新
💡 實務技巧:設施註冊本身不收費,但需確保美國代理人資訊正確且隨時可聯絡,否則 FDA 可能因無法聯繫而採取執法行動。
圖 2:FDA OTC 合規完整流程圖
此流程圖視覺化 FDA OTC 合規的四大核心步驟與時程規劃,幫助台灣業者建立完整的操作藍圖與時間表。
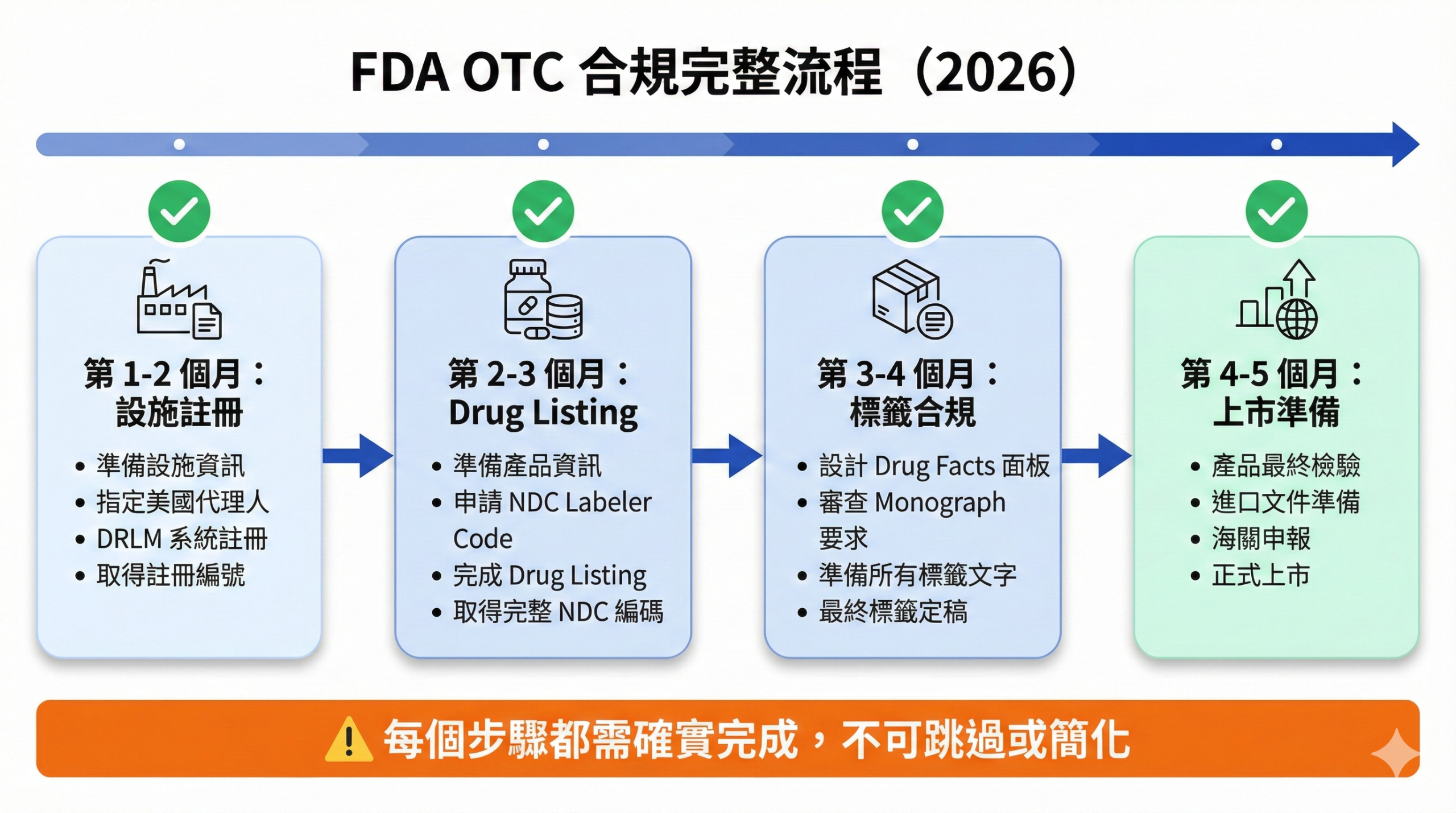
步驟二:Drug Listing(藥品列表申報)- 產品正式登記
什麼是 Drug Listing?
Drug Listing 是指將具體的 OTC 產品資訊(包含配方、劑型、包裝規格等)向 FDA 申報,並取得唯一的產品識別碼(NDC 編碼)。這是產品能否合法進入美國市場的關鍵步驟。
申報前的準備工作
在進行 Drug Listing 前,必須先完成:
- 設施註冊:沒有註冊編號無法進行 Listing
- NDC Labeler Code 申請:這是 NDC 編碼的前 5 碼,由 FDA 分配給責任方(Responsible Person)
- 產品配方確認:確保符合 OTC Monograph 或已取得 ANDA 核准
NDC 編碼結構解析
NDC(National Drug Code)是美國藥品的唯一識別碼,由 10 位數字組成:
| 部分 | 位數 | 說明 | 範例 |
|---|---|---|---|
| Labeler Code | 5碼 | FDA 分配給責任方的代碼 | 12345 |
| Product Code | 4碼 | 產品配方與劑型代碼(由責任方自訂) | 6789 |
| Package Code | 1碼 | 包裝規格代碼(由責任方自訂) | 0 |
完整 NDC 範例:12345-6789-0
⚠️ 重要提醒:Labeler Code 必須向 FDA 申請取得,不可自行編造。Product Code 與 Package Code 雖可自訂,但需遵循 FDA 編碼規則,且不同配方或包裝規格必須使用不同代碼。
Drug Listing 操作步驟
- 申請 Labeler Code:透過 DRLM 系統提交申請(首次申請約需 2-4 週)
- 準備產品資訊:
- 產品名稱(商品名 + 通用名)
- 活性成分與含量
- 劑型(片劑、軟膏、口服液等)
- 包裝規格與材質
- 產品類別(OTC Monograph 或 ANDA)
- 登入 DRLM 系統:選擇「Drug Listing」功能
- 填寫產品資訊:包含配方、劑型、包裝、標籤影本等
- 生成 NDC 編碼:系統會根據 Labeler Code 與自訂的 Product/Package Code 生成完整 NDC
- 提交 Listing:確認資訊無誤後提交
常見錯誤與避坑指南
| 錯誤類型 | 說明 | 正確做法 |
|---|---|---|
| 未申請 Labeler Code 就自訂 NDC | NDC 前 5 碼必須由 FDA 分配 | 先申請 Labeler Code 再進行 Listing |
| 不同包裝規格使用相同 Package Code | 每種包裝規格需有獨立 Package Code | 30 片裝與 60 片裝需使用不同 Package Code |
| 產品配方與 Monograph 不符 | Listing 會被拒絕或事後被要求下架 | 先確認符合 Monograph 再申報 |
| 未更新產品變更 | 配方或包裝變更需在 30 天內更新 Listing | 任何變更都需即時更新 DRLM 系統 |
💡 實務技巧:建議在產品設計階段就規劃好 NDC 編碼邏輯(如:不同劑量使用不同 Product Code、不同包裝使用不同 Package Code),避免後續混亂。
步驟三:標籤合規審查 – 最容易出錯的環節
Drug Facts 面板的強制要求
所有 OTC 藥品標籤都必須包含標準化的「Drug Facts」面板,這是 FDA 最嚴格審查的部分。面板內容包含:
- Active Ingredient(s):活性成分名稱與含量
- Purpose:產品用途(如:止痛、退燒)
- Uses:具體適應症
- Warnings:警語(包含禁忌、副作用、交互作用)
- Directions:使用方法與劑量
- Other Information:儲存條件等其他資訊
- Inactive Ingredients:非活性成分列表
標籤審查檢核清單
✅ 必須包含的資訊:
- Drug Facts 面板(格式與字體大小需符合 21 CFR Part 201.66)
- 責任方名稱與地址
- NDC 編碼(10 位數完整格式)
- 產品批號與有效期限
- 淨含量(需使用美制單位)
✅ 禁止的宣稱:
- 超出 Monograph 核准範圍的功效宣稱
- 暗示可治療嚴重疾病(如癌症、心臟病)
- 使用「天然」「有機」等可能誤導的用語(除非符合 FDA 定義)
❌ 常見錯誤:
- Drug Facts 面板字體過小(最小需 6 point)
- 警語不完整或翻譯錯誤
- 宣稱內容與 Monograph 不一致
- 未標示 NDC 編碼或格式錯誤
標籤審查流程
- 對照 Monograph 要求:確認所有必要資訊都已包含
- 檢查格式規範:字體大小、間距、排版符合 21 CFR Part 201.66
- 語言正確性:英文用詞專業且準確(建議由母語人士審查)
- 內部審查:品質部門與法規部門雙重確認
- 外部顧問審查:若資源允許,建議委託專業法規顧問最終審查
💡 實務技巧:許多 FDA 執法案例都源自標籤問題,建議在產品上市前進行「模擬 FDA 審查」,從嚴格的角度檢視所有標籤內容。
圖 3:FDA OTC 標籤合規檢核表
圖片用途說明:
此檢核表整理 OTC 標籤必須包含的所有元素與常見錯誤,幫助台灣業者在標籤設計階段就避免違規問題。

許多業者誤以為「完成註冊與 Listing 就萬事大吉」,實際上 FDA OTC 合規是持續性的義務,包含:
年度更新義務
- 設施註冊更新:每年 10-12 月必須更新
- Drug Listing 更新:產品資訊變更需在 30 天內更新
- NDC 編碼維護:停產或下架產品需通知 FDA
不良事件報告
若產品發生嚴重不良反應,責任方必須在 15 天內向 FDA 提交報告(21 CFR Part 310.305)。
cGMP 持續符合
製造設施必須持續符合 cGMP 要求,FDA 可隨時進行查廠。若發現違規,可能導致產品下架或設施被列入進口警示名單。
FDA 執法與市場監測
FDA 會透過市場監測、消費者投訴、進口查驗等方式持續監控 OTC 產品。若發現違規,可能採取:
- 警告信(Warning Letter)
- 產品扣押(Seizure)
- 進口禁令(Import Alert)
- 刑事起訴(嚴重違規情況)
🚨 法律風險:未完成註冊或 Listing 就銷售產品,將被視為「未經核准藥品」,責任方可能面臨每次違規最高 10 萬美元罰款。
需要特別注意的 3 個實務重點
1. 時程規劃要留足緩衝
建議在預計上市日期前 6 個月 開始啟動合規流程:
- 第 1-2 個月:設施註冊與 Labeler Code 申請
- 第 2-3 個月:Drug Listing 與 NDC 編碼取得
- 第 3-4 個月:標籤設計與審查
- 第 4-5 個月:產品試生產與最終檢驗
- 第 6 個月:進口與上市
2. 文件準備要完整且正確
常見的文件缺失包含:
- 設施註冊時未提供完整的美國代理人資訊
- Drug Listing 時產品配方描述不清
- 標籤影本未包含所有面板(正面、背面、側面)
- cGMP 文件不完整或翻譯錯誤
3. 責任方指定要明確
根據《FDA OTC 產品的角色、責任歸屬與法律風險判定:品牌與工廠誰會被連坐?》,責任方(Responsible Person)是承擔最終法律責任的主體。在合規流程中,所有註冊、Listing、標籤都必須以責任方名義進行,不可模糊不清。
總結
FDA OTC 合規實務路徑包含四大核心步驟,每個步驟都有明確的時程與文件要求:
- 設施註冊:取得入場資格,指定美國代理人
- Drug Listing:產品正式登記,取得 NDC 編碼
- 標籤合規:設計符合 FDA 規範的 Drug Facts 面板
- 上市後義務:年度更新、不良事件報告、cGMP 持續符合
建議在產品上市前 6 個月啟動合規流程,並確保每個步驟都完整且正確。任何環節的疏漏都可能導致產品無法上市或面臨 FDA 執法。
若對 FDA OTC 合規流程仍有疑問,歡迎參考 綠圈圈官網 的專業顧問服務,或閱讀我們的相關深度指南。