
你的產品會被視為醫療器械嗎?核心判定標準
是的,若產品預期用於診斷、治療、預防、緩解疾病,或影響人體結構/功能,即使未明確標示「醫療用途」,仍可能被 FDA 視為醫療器械(Medical Device)。 關鍵判定依據為「預期用途」(Intended Use),包含產品標籤、說明書、行銷宣稱,甚至網站描述。許多台灣外貿工廠與跨境賣家因不熟悉此標準,誤將醫療器械當作一般消費品出口,導致進口扣留或 FDA 警告信。
根據 21 CFR Part 860,FDA 將醫療器械分為 Class I(低風險)、Class II(中風險)、Class III(高風險)三類,不同類別對應不同合規要求。例如,體溫計屬 Class II,需完成 510(k) 審查或符合豁免條件;而心臟支架屬 Class III,需提交 PMA(上市前核准)申請。若產品分類錯誤或未完成必要申報,將面臨法律風險與商業損失。
本文將完整解析 FDA 醫療器械分類標準、台灣業者常見誤區、以及實務操作步驟,幫助您快速判定產品類別並確保合規出口。
為何台灣業者容易混淆醫療器械與一般產品?
台灣外貿工廠與跨境賣家常將「醫療器械」與「健康產品」「保健器材」混為一談,主要原因有三:
- 預期用途(Intended Use)定義模糊: FDA 判定標準不僅看產品本身,更看「如何宣稱使用」。例如,智能手環若宣稱「監測心率異常、預防心臟病」,即使未標註醫療用途,仍可能被視為 Class II 醫療器械。
- 台美法規差異: 台灣衛福部與 FDA 的醫療器械定義範圍不同。部分在台灣屬「一般商品」的產品(如某些按摩器、保健儀器),在美國可能需完成 FDA 註冊與 510(k) 審查。
- 代工廠責任認知不足: 許多 ODM/OEM 代工廠認為「合規是品牌商責任」,但根據 FDA 規定,製造商(Manufacturer)同樣需完成設施註冊(Establishment Registration)與器械列表申報(Device Listing),若品牌商違規,代工廠可能面臨連帶責任。
常見誤區範例:
- ❌ 「我們只是代工,不需要 FDA 註冊」→ 錯誤!製造商必須完成註冊
- ❌ 「產品未標註醫療用途,應該不算醫療器械」→ 錯誤!FDA 看預期用途,包含網站宣稱
- ❌ 「體溫計很簡單,不需要 510(k)」→ 錯誤!部分體溫計需 510(k),部分可豁免但仍需註冊
FDA 醫療器械三大分類與判定流程
Class I(一般管制):低風險器械
定義: 對人體風險最低,通常不涉及侵入性操作或直接接觸內臟。
常見產品: 手動輪椅、繃帶、手術手套、簡易體溫計(部分型號)、牙線
合規要求:
- 完成設施註冊(Establishment Registration)
- 完成器械列表申報(Device Listing)
- 多數產品豁免 510(k),但仍需符合 QSR(21 CFR Part 820)基本要求
- 需指定美國代理人(US Agent)
實務操作: 即使豁免 510(k),仍需每年更新註冊資訊,並確保產品標籤符合 FDA 要求(包含 UDI、製造商資訊、使用說明)。
Class II(特別管制):中風險器械
定義: 對人體有中等風險,需額外管制措施確保安全有效性。
常見產品: 電子體溫計、血壓計、X 光機、助聽器、某些診斷試劑、外科手術器械
合規要求:
- 完成設施註冊與器械列表申報
- 多數需提交 510(k) Premarket Notification(上市前通知),證明產品與已核准的同類產品(Predicate Device)具實質等效性(Substantial Equivalence)
- 部分產品可豁免 510(k),但需符合特定條件(參考 FDA 豁免清單)
- 符合 QSR 全面要求
- 需指定美國代理人
實務操作: 510(k) 審查時間通常 90-180 天,需準備技術文件、臨床數據(若需要)、與 Predicate Device 的比較分析。若無合適 Predicate Device,需考慮 De Novo 途徑。
Class III(上市前核准):高風險器械
定義: 對人體有高風險,通常涉及生命維持、侵入性操作或植入體內。
常見產品: 心臟支架、人工心臟瓣膜、植入式心律調節器、某些體外診斷設備(IVD)
合規要求:
- 完成設施註冊與器械列表申報
- 必須提交 PMA(Premarket Approval)申請,包含完整臨床試驗數據證明安全性與有效性
- 符合 QSR 全面要求
- 需指定美國代理人
- PMA 審查時間通常 180-360 天或更長
實務操作: PMA 申請成本高、審查嚴格,需提供臨床試驗數據、製造流程文件、品質管理系統證明。台灣業者若涉及 Class III 產品,建議尋求專業法規顧問協助。
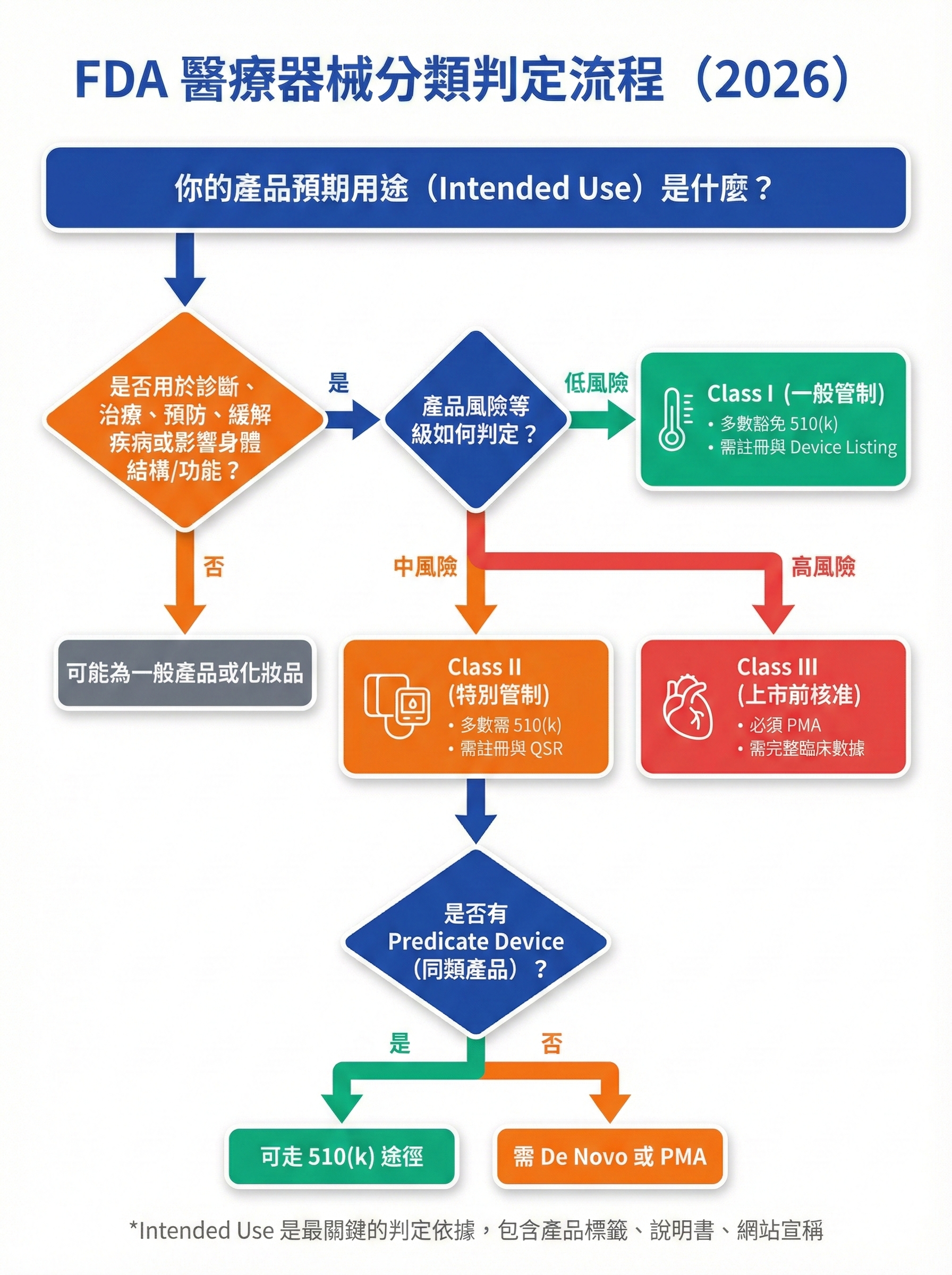
圖 2: FDA 醫療器械分類判定決策樹
圖片用途說明:
此決策樹將「產品是否被視為醫療器械」及「屬於哪一類」的判定邏輯視覺化,幫助台灣賣家與外貿工廠快速自我檢測產品分類與合規路徑。
台灣業者最常踩錯的 6 個合規紅線
紅線 1: 產品宣稱醫療用途但未完成 FDA 註冊
情境: 跨境賣家在 Amazon 或自有網站宣稱產品「監測血糖」「預防高血壓」,但未完成 FDA 設施註冊與器械列表申報。
風險: FDA 可發出警告信(Warning Letter)、進口扣留、甚至民事罰款。品牌商與製造商(代工廠)均可能被追究責任。
避險策略: 若產品涉及任何醫療宣稱,務必先完成 FDA 註冊。即使是代工廠,也需以製造商身分註冊。
紅線 2: 未提交 510(k) 或誤判豁免條件
情境: 業者認為「簡單的醫療器械」不需 510(k),但實際上該產品不符合豁免清單條件。
風險: 未經核准即上市,屬違法行為,可能面臨產品召回、罰款、甚至刑事責任。
避險策略: 查閱 FDA 官方豁免清單(21 CFR Part 862-892),不確定時諮詢專業顧問。即使豁免 510(k),仍需完成註冊與 Device Listing。
紅線 3: 標籤未包含必要資訊(UDI、製造商、使用說明)
情境: 產品標籤僅標註品牌名稱與型號,未包含 UDI(唯一器械識別碼)、製造商資訊、使用說明、警告事項。
風險: 違反 21 CFR Part 801 標籤要求,可能導致進口扣留或 FDA 警告信。
避險策略: 確保標籤包含:
- UDI(符合 GUDID 資料庫格式)
- 製造商名稱與地址
- 美國代理人資訊
- 使用說明與警告事項
- 必要時標註「Rx Only」(處方器械)或「OTC」(非處方器械)
紅線 4: 未完成設施註冊或資訊過期
情境: 代工廠完成初次註冊後,未每年更新註冊資訊(每年 10 月 1 日至 12 月 31 日為更新期限)。
風險: 註冊失效,產品無法合法進口美國。
避險策略: 設定提醒,每年更新註冊資訊。若設施地址、負責人、產品線有變更,需立即更新。
紅線 5: 未指定美國代理人或資訊錯誤
情境: 業者未指定美國代理人(US Agent),或代理人資訊已過期但未更新。
風險: 違反 21 CFR Part 807.40 要求,FDA 無法聯繫業者,可能導致進口扣留。
避險策略: 指定可靠的美國代理人(可為個人或公司),確保代理人資訊正確且隨時可聯繫。若代理人變更,需立即向 FDA 更新。
紅線 6: 製造設施未符合 QSR(21 CFR Part 820)要求
情境: 代工廠未建立品質管理系統(QMS),或 QMS 文件不完整、未定期稽核。
風險: FDA 可進行突擊檢查(Inspection),若發現不符合 QSR 要求,可發出 Form 483(缺失報告)或警告信。嚴重者可能被列入進口警示清單(Import Alert)。
避險策略: 建立完整 QMS,包含:
- 設計管制(Design Controls)
- 文件管制(Document Controls)
- 採購管制(Purchasing Controls)
- 製程驗證(Process Validation)
- 不良事件報告(Adverse Event Reporting)
- 定期內部稽核與管理審查
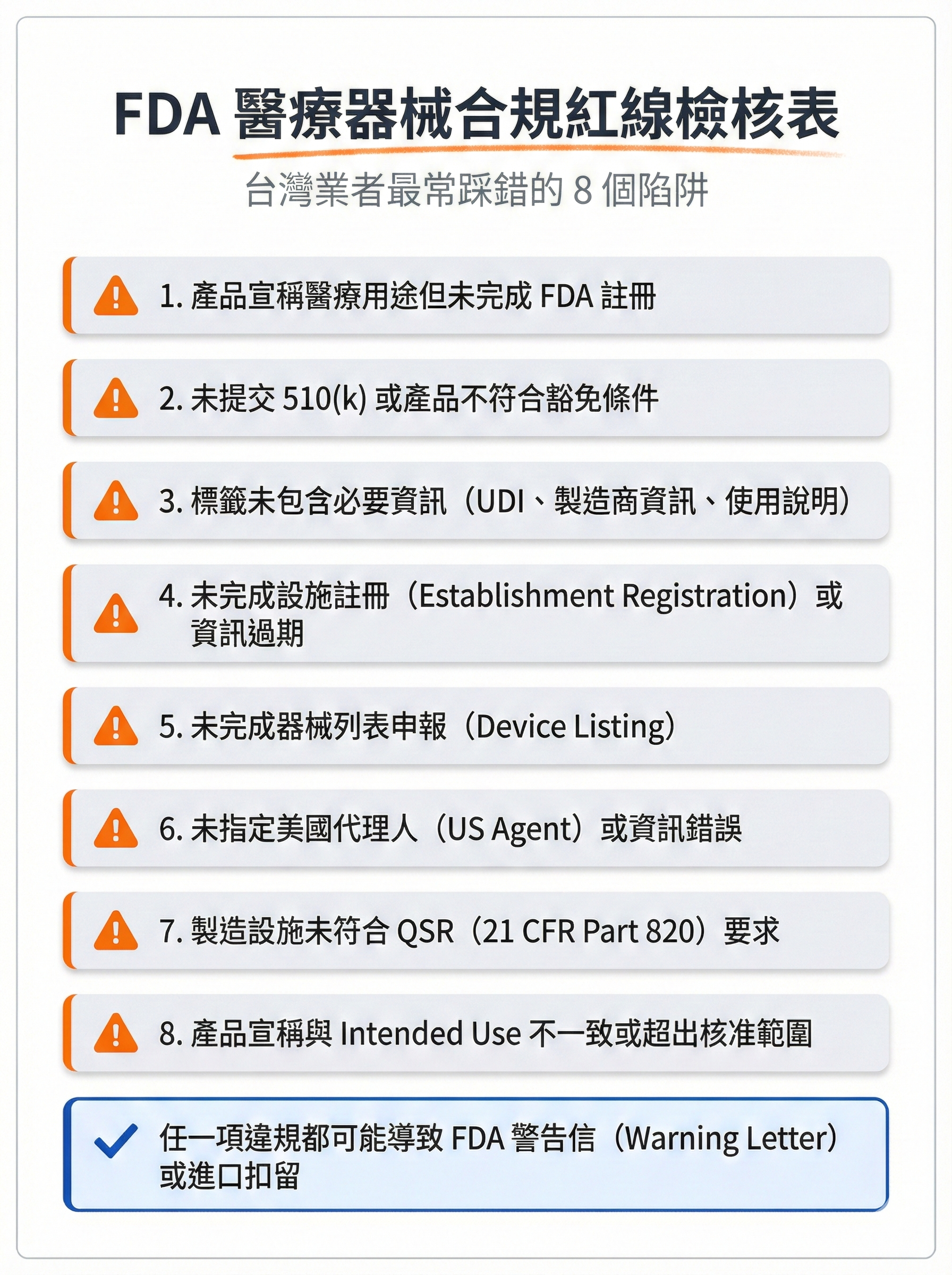
圖 3: FDA 醫療器械合規紅線檢核表
圖片用途說明:
此檢核表整理台灣賣家與外貿工廠最常踩錯的 8 個醫療器械合規紅線,幫助業者快速自我檢查,避免違規風險與商業損失。
實務操作:台灣業者如何完成 FDA 合規?
步驟 1: 判定產品是否為醫療器械
操作: 檢視產品預期用途(Intended Use),包含:
- 產品標籤與說明書
- 網站宣稱與行銷文案
- 產品功能與使用情境
判定標準: 若涉及診斷、治療、預防、緩解疾病或影響身體結構/功能,即為醫療器械。
常見誤區: 即使產品未明確標註「醫療用途」,若網站宣稱「改善血液循環」「預防疾病」,仍可能被視為醫療器械。
步驟 2: 確認產品分類(Class I/II/III)
操作: 查閱 FDA 產品分類資料庫(Product Classification Database),輸入產品名稱或關鍵詞,查詢對應分類代碼(Product Code)與管制類別。
範例:
- 電子體溫計: Product Code FLL, Class II
- 血壓計: Product Code DXN, Class II
- 手動輪椅: Product Code IOR, Class I
注意: 同類產品可能因設計、用途不同而屬不同分類。例如,臨床用體溫計(Class II)與家用體溫計(部分 Class I)。
步驟 3: 完成設施註冊與器械列表申報
操作: 透過 FDA 電子系統(FURLS)完成:
- Establishment Registration(設施註冊): 提供製造商名稱、地址、負責人資訊
- Device Listing(器械列表申報): 提供產品名稱、型號、分類代碼、預期用途
時程: 初次註冊需 5-10 個工作天,每年 10-12 月需更新註冊資訊。
費用: 設施註冊與 Device Listing 本身免費,但若需提交 510(k) 或 PMA,需支付使用者費用(MDUFA)。
步驟 4: 評估是否需提交 510(k) 或 PMA
Class I: 多數豁免 510(k),僅需完成註冊與 Device Listing。
Class II: 多數需提交 510(k),部分產品可豁免(查閱豁免清單)。
Class III: 必須提交 PMA,包含完整臨床試驗數據。
510(k) 準備文件:
- 產品技術規格與設計文件
- 與 Predicate Device 的比較分析(Substantial Equivalence)
- 性能測試數據(如生物相容性測試、電氣安全測試)
- 標籤與使用說明書
- 製造流程與 QMS 文件
步驟 5: 建立品質管理系統(QMS)
操作: 依據 21 CFR Part 820(QSR)建立 QMS,包含:
- 設計管制(Design Controls)
- 文件管制(Document Controls)
- 採購管制(Purchasing Controls)
- 製程驗證(Process Validation)
- 不良事件報告(Adverse Event Reporting,MDR)
- 定期內部稽核與管理審查
建議: 可參考 ISO 13485 標準(國際醫療器械品質管理系統),有助於同時符合 FDA 與其他市場(如歐盟 CE Mark)要求。
步驟 6: 指定美國代理人並完成產品標籤
美國代理人: 需為美國境內個人或公司,負責接收 FDA 通知、協助溝通。
產品標籤: 確保包含:
- UDI(唯一器械識別碼,需註冊至 GUDID 資料庫)
- 製造商名稱與地址
- 美國代理人資訊
- 使用說明與警告事項
- 必要時標註「Rx Only」或「OTC」
需要特別注意的 3 個重點
重點 1: 代工廠與品牌商的責任歸屬
根據 FDA 規定,製造商(Manufacturer)與品牌商(Labeler/Distributor)均需完成註冊。若品牌商違規,代工廠可能面臨連帶責任。建議在合約中明確約定:
- 誰負責 FDA 註冊與 510(k) 申請
- 誰負責產品標籤與宣稱內容
- 違規責任如何分攤
重點 2: 醫療器械 vs 體外診斷設備(IVD)
部分產品(如血糖試紙、COVID-19 快篩)屬體外診斷設備(In Vitro Diagnostic Devices, IVD),適用 21 CFR Part 809-866,管制要求與一般醫療器械略有不同。若產品涉及實驗室檢測或診斷,需確認是否屬 IVD。
重點 3: FDA 與 CE Mark 的差異
台灣業者若同時出口美國與歐盟,需注意:
- FDA: 強調上市前審查(510(k)/PMA)與製造商註冊
- CE Mark: 強調符合性聲明(Declaration of Conformity)與技術文件(Technical Documentation)
- 兩者可互補: 符合 ISO 13485 有助於同時滿足 FDA QSR 與歐盟 MDR 要求
總結
- ✅ 產品是否為醫療器械,取決於「預期用途」(Intended Use),包含標籤、說明書、網站宣稱
- ✅ FDA 將醫療器械分為 Class I/II/III,不同類別對應不同合規要求(註冊、510(k)、PMA)
- ✅ 台灣代工廠與品牌商均需完成 FDA 註冊,代工廠不可誤認「合規是品牌商責任」
- ✅ 最常見的合規紅線包含: 未完成註冊、未提交 510(k)、標籤不符、QSR 不合格
- ✅ 建議建立完整 QMS(符合 QSR 或 ISO 13485),有助於通過 FDA 檢查並拓展其他市場
若對 FDA 醫療器械合規仍有疑問,或需協助判定產品分類、準備 510(k) 文件,歡迎參考 綠圈圈官網 的專業顧問服務。